Diagnosis
Cystic fibrosis FAQs
Pulmonologist Sarah Chalmers, M.D., answers the most frequently asked questions about cystic fibrosis.
Hello. I'm Dr. Sarah Chalmers, a pulmonologist at Mayo Clinic. And I'm here to answer some of the important questions you may have about cystic fibrosis.
Just because your baby's newborn screen came back positive does not mean that your baby has cystic fibrosis. Most babies who have a positive screening actually don't have CF. The newborn screen looks at a substance in the blood that is elevated in cystic fibrosis, but it can be elevated in other conditions as well, even premature birth. Some states also test for a gene mutation, but even if this comes back positive, it doesn't mean your baby has the disease. People with only one mutation are called carriers. It's very common in the United States and one in 20 people are CF gene mutation carriers. If your baby has a positive cystic fibrosis screen, they will need to see their doctor and have a sweat chloride test to see if they do have cystic fibrosis.
CF gene mutations are actually passed from parent to children in a specific pattern called autosomal recessive. Each parent passes one CF gene to their child, and therefore each person has two CF genes. To get the disease, both genes have to have a mutation. People with one CF gene are called carriers. If a parent is a carrier, there's a 50 percent chance they'll pass on the gene with a mutation to their child. If both parents pass on a normal gene, or only one parent passes a gene with a mutation, the child will not have CF. If both parents pass on a gene with a mutation, then the baby will have two genes with the mutation and will likely get the disease. If both parents are CF mutation carriers, there's a 25 percent chance that each one of their babies will be born with cystic fibrosis.
So both males and females can get cystic fibrosis. But females tend to have more symptoms, more lung infections, and they tend to start these symptoms of infections earlier in life as compared to males. No one knows for sure why this is so.
Actually, nearly 10 percent of cases of CF are diagnosed in adulthood. You're born with cystic fibrosis, but there are several reasons why it may not be diagnosed during childhood. Prior to 2010, some states didn't even screen for cystic fibrosis. So if you were born before 2010, you may not have received a newborn screening test for cystic fibrosis as a baby. Some gene mutations cause very mild disease and symptoms may go unnoticed until adulthood.
CF symptoms, how the disease affects the patient's organs and how it impacts their life is very different from one person to the next. Some people have very mild disease with only one organ affected and very few symptoms, while others have more severe disease with troublesome symptoms and multiple organs that are affected. Many factors including gene mutation type determine the impact on the patient. But your cystic fibrosis care team can work with you as an individual patient to create a personalized treatment plan that meets your individual needs.
Fertility is affected in both men and women with cystic fibrosis. Women with CF have thicker cervical mucus and they may also have irregular menstrual cycles. So it may take longer for women with CF to become pregnant. But most can become pregnant, have a normal pregnancy and a normal delivery. Almost all men with CF have infertility. Men with CF make normal sperm, but the sperm canal is absent. Because they still make sperm, assisted reproductive technologies can be used to help male CF patients have biologic children. Whether your children get CF or not depends on the combination of genes passed on from you and your significant other and can range from zero chance if neither parent has a gene mutation to a near 100% chance if both parents have CF.
Always be honest with your health care team. Let us know which medications you're taking and how often you're doing your treatments. Write down your questions before you come to your appointment so that we can make sure that we are meeting your needs. Thanks for your time. And we wish you well.
To diagnose cystic fibrosis, healthcare professionals typically do a physical exam, review your symptoms and do tests.
Newborn screening and diagnosis
Every state in the U.S. now routinely screens newborns for cystic fibrosis. Early diagnosis means that treatment can begin right away. Testing can include:
- Newborn screening. In this screening test, a healthcare professional takes a few drops of blood from the baby's heel. A lab checks the blood sample for higher levels than expected of a chemical called immunoreactive trypsinogen (IRT). IRT is released by the pancreas and may suggest CF. A newborn's IRT levels also may be high because of premature birth or a stressful delivery. For that reason, other tests may be needed to confirm a diagnosis of cystic fibrosis.
- Sweat test. To check if a baby has CF, a sweat test is done once the baby is at least 2 weeks old. A chemical that causes the skin to sweat is put on a small area of skin. Then the sweat is collected to test it and see if it's saltier than typical. Testing done at a care center accredited by the Cystic Fibrosis Foundation helps ensure results that can be trusted.
- Genetic testing. Healthcare professionals also may recommend genetic testing to look for specific changes on the gene responsible for CF. Genetic testing may be used along with IRT levels to confirm the diagnosis.
Testing of older children and adults
Cystic fibrosis tests may be recommended for older children and adults who weren't screened at birth. Your healthcare professional may suggest genetic and sweat tests for CF if you have repeated bouts of an inflamed pancreas, nasal polyps, chronic sinus infections, lung infections, bronchiectasis or male infertility.
More Information
Treatment
There is no cure for cystic fibrosis, but treatment can ease symptoms, lessen complications and improve quality of life. Close monitoring and early, aggressive intervention is recommended to slow the worsening of CF over time. This can lead to a longer life.
Managing CF is complicated, so it's best to get treatment at a center with a multispecialty team of doctors and other healthcare professionals trained in CF. They can evaluate and treat your condition.
The goals of treatment include:
- Preventing and controlling infections that occur in the lungs.
- Removing and loosening mucus from the lungs.
- Treating and preventing intestinal blockage.
- Getting enough nutrition.
Medicines
Options include:
- Medicines that target gene changes and improve how the CFTR protein works. These are called cystic fibrosis transmembrane conductance regulator (CTFR) modulators.
- Antibiotics to treat and prevent lung infections.
- Anti-inflammatory medicines to lessen swelling in the airways in the lungs.
- Mucus-thinning medicines, such as hypertonic saline, to help cough up mucus. This can improve lung function.
- Medicines breathed into the lungs called bronchodilators. These can help keep airways open by relaxing the muscles around the bronchial tubes.
- Pancreatic enzyme capsules taken by mouth to help the digestive tract take in and use nutrients.
- Stool softeners to prevent constipation or bowel obstruction.
- Acid-reducing medicines to help pancreatic enzymes work better.
- Specific medicines for diabetes or liver disease, when needed.
Medicines that target genes
For those with cystic fibrosis who have certain gene changes, cystic fibrosis transmembrane conductance regulator (CFTR) modulators may help. About 90% of people with CF may be helped by using these medicines. Gene testing is needed to find out which specific gene change you have and if a CFTR modulator may work for you.
CFTR modulators are newer medicines that many experts think are a breakthrough in the treatment of CF. The medicines help the CFTR protein work better. This can make lung function better, help digestion and weight, and lessen the amount of salt in sweat.
The U.S. Food and Drug Administration (FDA) has approved these CFTR modulators for treating CF in people with specific changes in the CFTR gene:
- The newest combination medicine with elexacaftor, ivacaftor and tezacaftor (Trikafta) is approved for people age 2 years and older. Trikafta has been shown to be the most effective CFTR modulator.
- The combination medicine with ivacaftor and tezacaftor (Symdeko) is approved for people age 6 years and older.
- The combination medicine with ivacaftor and lumacaftor (Orkambi) is approved for people who are age 1 year and older.
- Ivacaftor (Kalydeco) is approved for people who are 1 month and older.
Your healthcare professional may do liver function tests and eye exams before prescribing these medicines. While taking these medicines, you'll likely need testing on a regular basis to check for side effects such as liver function changes and clouding of the eye lenses called cataracts. Ask your healthcare professional and pharmacist for information on possible side effects and what to watch for.
Keep regular follow-up appointments so your healthcare professional can monitor you while taking these medicines. Tell your healthcare professional about any side effects that you have.
Airway clearance techniques
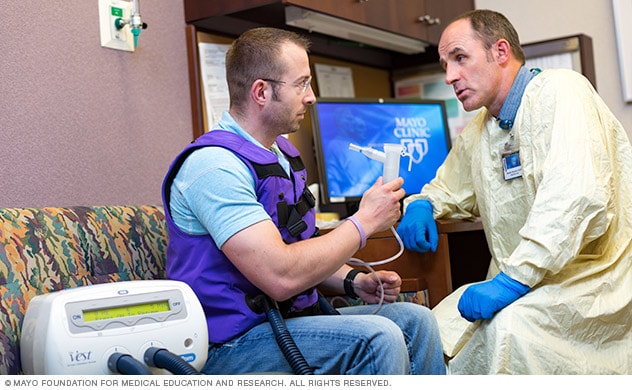 Vest therapy
Vest therapy
Using a personalized approach, a Mayo Clinic respiratory therapist discusses inflatable vest therapy with an adult who has cystic fibrosis.
Airway clearance techniques, also called chest physical therapy, can help get rid of mucus blocking the airways. It also can help to lessen infection and inflammation in the airways. Airway clearance techniques loosen the thick mucus in the lungs, making it easier to cough up.
Airway clearing techniques are usually done several times a day. Different techniques, and often more than one method, can be used to loosen and remove mucus.
- Clapping with cupped hands on the front and back of the chest. This is a common technique.
- Special breathing and coughing activities.
- Mechanical devices, such as a tube that you blow into, and a machine that pulses air into the lungs called a vibrating vest.
- Vigorous exercise.
Your healthcare professional can give you instructions on the airway clearance techniques that are best for you and how often you should do them.
Pulmonary rehabilitation
Your healthcare professional may recommend a long-term program called pulmonary rehabilitation. The program may improve your lung function and your overall well-being. Pulmonary rehabilitation is usually done on an outpatient basis and may include:
- Physical exercise that may improve your condition.
- Breathing techniques that may help loosen mucus and make breathing easier.
- Dietary counseling.
- Mental health counseling and support.
- Education about your condition.
Surgery and other treatments
Options for certain conditions caused by cystic fibrosis include:
- Nasal and sinus surgery. Surgery can remove nasal polyps that get in the way of breathing. Sinus surgery may be done to treat repeated or long-term sinusitis.
- Oxygen therapy. If there isn't enough oxygen in your blood, you may need supplemental oxygen. You can get this extra oxygen to your lungs through a mask or through plastic tubing with tips that fit into your nose. These attach to an oxygen tank. Lightweight, portable units that you take with you can help you be more mobile. Oxygen therapy may prevent high blood pressure in the lungs, a condition called pulmonary hypertension.
- Noninvasive ventilation. Typically used while sleeping, noninvasive ventilation uses a nose or mouth mask to give positive pressure in the airway and lungs when breathing in. It's often used along with oxygen therapy. Noninvasive ventilation can increase air exchange in the lungs and lessen the work of breathing. The treatment also may help with airway clearance.
- Feeding tube. CF interferes with digestion, so you can't take in and use nutrients from food very well. A feeding tube delivers extra nutrition. This may be a short-term tube placed through your nose and guided to your stomach. Or the tube may be surgically placed in the stomach through a small cut in the skin on your belly. A feeding tube gives extra calories during the day or night and does not keep you from eating by mouth.
- Bowel surgery. If a blockage happens in the intestines, you may need surgery to remove it. If part of an intestine folds inside a nearby section of intestine, you may need surgery.
-
Lung transplant. If you have severe breathing problems or life-threatening lung complications, or if antibiotics no longer work to treat lung infections, a lung transplant may be an option. Because bacteria line the airways in diseases such as CF that cause permanent widening of the large airways, both lungs need to be replaced.
Cystic fibrosis does not recur in transplanted lungs. But other complications linked with CF, such as sinus infections, diabetes, pancreas conditions and osteoporosis, can still happen after a lung transplant.
- Liver transplant. For severe CF-related liver disease, such as cirrhosis, liver transplant may be an option. In some people, a liver transplant may be done together with lung or pancreas transplants.
More Information
Clinical trials
Explore Mayo Clinic studies testing new treatments, interventions and tests as a means to prevent, detect, treat or manage this condition.
Lifestyle and home remedies
Here are some ways you can manage cystic fibrosis and lessen complications.
Pay attention to nutrition and fluid intake
Cystic fibrosis can cause poor nutrition because the enzymes needed for digestion can't reach the small intestine. This prevents food from being taken in and used by the body. People with CF may need a much higher number of calories daily than do people without the condition.
A healthy diet is important to growth and development and to support good lung function. It's also important to drink lots of fluids to help thin the mucus in your lungs. You may work with a dietitian to create a nutrition plan.
Your healthcare professional may recommend:
- Pancreatic enzyme capsules with every meal and snack.
- Medicines to lessen acid made in the stomach and help pancreatic enzymes work.
- High-calorie nutrition supplements.
- Special fat-soluble vitamins.
- Extra fiber to prevent intestinal blockage.
- Extra salt, especially during hot weather or before exercising.
- Drinking enough water, especially during hot weather.
Keep vaccinations up to date
In addition to the other usual childhood vaccines, the annual flu vaccine is important if you have cystic fibrosis. So are any other vaccines your healthcare professionals recommend, such as the vaccine to prevent pneumonia and COVID-19. CF doesn't affect the immune system, but people with CF are more likely to develop complications when they get sick.
Exercise
Regular exercise helps loosen mucus in your airways and makes your heart stronger. Because people with cystic fibrosis are living longer, it's important to keep your heart and blood vessels in good shape for a healthier life. Anything that gets you moving, including walking and biking, can help.
Stay away from smoke
Don't smoke, and don't allow other people to smoke around you or your child. Secondhand smoke and air pollution are harmful for everyone, but especially if you have cystic fibrosis. Using electronic cigarettes, also called vaping, can worsen CF too.
Wash your hands
Teach all the members of your family to wash their hands thoroughly before eating, after using the bathroom, when coming home from work or school, and after being around a sick person. If possible, stay away from people who have colds or flu. Washing your hands is the best way to protect against infection.
Keep medical appointments
Along with ongoing care from your medical team:
- Keep your regular follow-up appointments.
- Take your medicines as prescribed and follow therapies as instructed.
- Talk with your healthcare professional about how to manage symptoms.
- Learn the warning signs of serious complications.
Coping and support
If you or someone you love has cystic fibrosis, you may have strong emotions such as depression, anxiety, anger or fear. These feelings may be especially common in teens. These tips may help.
- Find support. Talking openly about how you feel can help. It also may help to talk with others who have the same condition. That might mean joining a support group for yourself or finding a support group for parents of children with cystic fibrosis. Older children with CF may want to join a CF group to meet and talk with others who have the condition.
- Get professional help. If you or your child is depressed or anxious, it may help to meet with a mental health professional. You can talk about feelings and ways to cope. The mental health professional may suggest medicines or other treatments too.
- Spend time with friends and family. Having their support can help you manage stress and lessen anxiety. Ask your friends or family for help when you need it.
- Take time to learn about cystic fibrosis. If your child has cystic fibrosis, encourage your child to learn about CF. Find out how medical care is managed for children with CF as they grow older and reach adulthood. Talk with your healthcare professional if you have questions about care.
Preparing for your appointment
Make an appointment with your healthcare professional if you or your child has symptoms common to cystic fibrosis. After the evaluation, you may be referred to a specialist trained in diagnosing and treating CF.
Here's some information to help you prepare for your appointment, as well as what to expect from your healthcare professional.
What you can do
You might want to take a friend or family member with you to the appointment to help you remember information.
Before your appointment, make a list of:
- Symptoms and when they started. Include anything that makes symptoms worse or better.
- All medicines, vitamins, herbs and supplements that you or your child take. Include the doses.
- Family history, such as whether anyone in your family has cystic fibrosis.
- Treatment you or your child have had for CF, if any. Include what the treatment was and if it helped.
- Any other medical conditions and their treatments.
- Questions to ask your healthcare professional.
Questions to ask may include:
- What is likely causing these symptoms?
- What kinds of tests are needed?
- What treatment do you recommend?
- I or my child have other health conditions. How will cystic fibrosis affect them?
- Are there any limits needed?
Feel free to ask other questions during your appointment.
What to expect from your doctor
After getting detailed information about the symptoms and your family's medical history, your healthcare professional may order tests to help with diagnosis and plan treatment.
Your healthcare professional also may ask questions, such as:
- What symptoms are you or your child having?
- When did the symptoms start?
- Does anything make the symptoms better or worse?
- Has anyone in your family ever had cystic fibrosis?
- Has growth been average and weight been stable?
Dec. 07, 2024