Oct. 18, 2019
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic interstitial lung disease with a median survival of 2.5 to 5 years. Until 2014, lung transplantation was the only available treatment ― along with supportive therapies including oxygen, pulmonary rehabilitation, and the management of comorbidities such as obstructive sleep apnea or symptomatic gastroesophageal reflux disease.
Current medical management involves two oral anti-fibrotic agents: nintedanib and pirfenidone. Nintedanib is a triple tyrosine kinase inhibitor acting at multiple sites in the fibrotic pathway, while the exact mechanism of pirfenidone remains unknown. "Both agents demonstrate comparable slowing of functional decline as measured by change in forced vital capacity (FVC) on pulmonary function testing over a 12-month study period," says Teng Moua, M.D., a pulmonologist at Mayo Clinic's campus in Rochester, Minnesota. "Neither has demonstrated significant influence on patient-reported outcomes such as dyspnea or respiratory-related quality of life. Both are similar in cost, with currently limited data on long-term survival or mortality improvement."
Results vary with treatment options for IPF
Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis
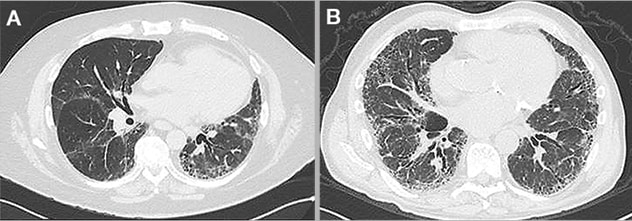
Computed tomography (CT) examples of two patients with suspected idiopathic pulmonary fibrosis. Patient (A) has a probable usual interstitial pneumonia pattern. Patient (B) has a more consistent pattern. Diagnostic confidence leading to treatment is often greater in patients with more consistent radiologic patterns compared to those with probable or indeterminate findings, which may delay treatment in the latter. In these two patients, anti-fibrotic therapy was started immediately at presentation, despite diagnostic uncertainty in the first patient.
While widely available, a survey study of European practices published in BMC Pulmonary Medicine in 2017 suggested up to 40% of diagnosed patients with IPF remain untreated. Barriers to drug initiation include:
- Uncertainty with atypical presentations, particularly those with earlier or inconsistent radiologic findings
- Concern for untoward side effects in those with more stable or slowly progressive disease
- Lack of perceived clinical benefit in asymptomatic patients, or those with normal or already severely limited pulmonary function testing.
Drug effect on rate of FVC decline also appears variable as reviewed in a manuscript published in the American Journal of Respiratory and Critical Care Medicine in 2016 suggesting early stabilization of pre-treatment decline within three months of treatment initiation while in some there may not be appreciable change for a year or more.
Results of a large clinical trial involving pirfenidone published in the New England Journal of Medicine in 2014 found close to 20% of those in the treatment arm saw a clinically important decline in FVC (greater than 10% over the 12-month study period), suggesting some patients may not have an early response to directed treatment or none at all. For these and other reasons, many clinicians and patients have taken a conservative approach to starting anti-fibrotic therapy.
Researchers have attempted to study which subgroups of IPF patients would continue to benefit from real-world treatment outside of clinical trials. Subsequent reports are derived from post-hoc analyses of large clinical trials or smaller single-center observational cohorts and suggest functional response in a number of patients with concerning features.
For example, improvement in FVC decline in those with more-consistent versus probable or suggestive radiologic findings was similar in one post-hoc analysis published in the American Journal of Respiratory and Critical Care Medicine in 2017. Rate of FVC decline on drug therapy appeared similar in those with severe disease (defined as FVC less than 50% of predicted) to those with mild or moderate disease. Patients with normal (more than 70% or 90%) FVC at presentation also had similar rates of decline when initiated on therapy.
"As respiratory symptoms are not alleviated by anti-fibrotic therapy, patients who are asymptomatic may find it difficult to initiate and sustain treatment due to imposed adverse effects. There is no systematic data to suggest onset of symptoms would be delayed in asymptomatic patients with or without PFT abnormality," says Dr. Moua. "Patients with combined pulmonary fibrosis and emphysema, whose FVC decline may be difficult to follow due to false normalization, remain a challenging subgroup to treat."
Radiologic emphysema may occur in up to 10% of patients with IPF, with the primary controlled trial, published in the New England Journal of Medicine in 2014, specifically excluding any patient with radiologic emphysema from trial participation and subsequently limiting the systematic evaluation of anti-fibrotic therapy in such patients.
Continued research needed for anti-fibrotics therapy
Anti-fibrotics are currently being studied in other non-IPF progressive interstitial lung diseases. An international trial found nintedanib to be effective in slowing FVC decline in patients with scleroderma-related lung fibrosis. Treatment medication was overall well-tolerated with only about a 16% withdrawal or discontinuation rate. The study appeared in the New England Journal of Medicine in 2019.
Findings from an international study looking at nintedanib for the treatment of progressive non-IPF fibrotic lung disease, published in the New England Journal of Medicine in 2019, noted an improved rate of overall disease decline in those patients on therapy versus placebo. Disease progression in the study was defined by specific clinical, radiologic, and functional changes. The study ultimately enrolled fibrotic subtypes included chronic hypersensitivity pneumonitis, connective-tissue disease-related interstitial lung disease, occupational, and fibrotic sarcoidosis. Patients already on traditional treatments (corticosteroids, steroid-sparing agents) were allowed to keep such therapies in addition to trial nintedanib.
Similarly, a phase II double-blind randomized controlled trial found pirfenidone to be well tolerated in progressive -unclassified diseases, but did not successfully model change in its primary outcome of FVC decline as measured by daily home spirometry. Site spirometry testing did suggest decreased rate of FVC decline among those taking trial drug. Study results were published in Lancet Respiratory Medicine in 2019.
An ongoing trial looking at the efficacy of pirfenidone in rheumatoid arthritis-related fibrotic lung disease, TRAIL-1, is actively recruiting across multiple sites in the United States.
While ushering in an era of medical treatment for a once-progressive and fatal disease, caveats to management in certain IPF subgroups and the expansion of indications to include other non-IPF progressive fibrotic lung disease highlight the current state of the available anti-fibrotics.
For more information
Maher TM, et al. Unmet needs in the treatment of idiopathic pulmonary fibrosis―insights from patient chart review in five European countries. BMC Pulmonary Medicine. 2017;17:124.
Costabel U, et al. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in INPULSIS. American Journal of Respiratory and Critical Care Medicine. 2016;193:178.
King TE, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. New England Journal of Medicine. 2014;370:2083.
Raghu G, et al. Effect of nintedanib in subgroups of idiopathic pulmonary fibrosis by diagnostic criteria. American Journal of Respiratory and Critical Care Medicine. 2017;195:78.
Distler O, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. New England Journal of Medicine. 2019;380:2518.
Flaherty KR, et al. Nintedanib in progressive fibrosing interstitial lung diseases. New England Journal of Medicine. In press.
Maher TM, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: A double-blind, randomized, placebo controlled, phase 2 trial. Lancet Respiratory Medicine. In press.