June 20, 2024
Polycystic kidney disease (PKD) is an inherited disorder in which clusters of fluid-filled cysts develop primarily within the kidneys, causing the kidneys to enlarge and lose function over time. Polycystic kidney disease also can cause cysts to develop in the liver and elsewhere in the body. The disease can cause serious complications, including high blood pressure and kidney failure.
"Mayo Clinic has the multidisciplinary expertise and collaborative culture necessary for diagnosing and treating PKD," says Christian Hanna, M.D., M.S., a pediatric nephrologist at Mayo Clinic Children's Center in Rochester, Minnesota. "Our team — including nephrologists, hypertension specialists, geneticists, neurologists, surgeons and radiologists — works together to develop a comprehensive, personalized treatment plan," says Dr. Hanna.
Mayo Clinic doctors and research scientists specializing in PKD collaborate to find new approaches, offering multidisciplinary expertise, personalized care, advanced diagnostics, improved prognostics, innovative therapies and access to clinical trials.
Evaluation, diagnosis and personalized treatment plans
The two main types of polycystic kidney disease, caused by different genetic flaws, are:
- Autosomal dominant polycystic kidney disease (ADPKD). Symptoms of ADPKD often develop between the ages of 30 and 40. By age 60, approximately half of the patients with PKD will have developed kidney failure. Although this type was historically referred to as adult polycystic kidney disease, children also can develop the disease and experience symptoms such as high blood pressure, abdominal or back pain, and blood in the urine. Only one parent needs to have the disease for it to be passed on to the children. If one parent has ADPKD, each child has a 50% chance of inheriting the disease. ADPKD accounts for most cases of polycystic kidney disease.
- Autosomal recessive polycystic kidney disease (ARPKD). This type is less common than ADPKD. Symptoms typically appear shortly after birth, although sometimes they may not manifest until later in childhood or during adolescence. Children born with this condition often develop kidney failure before reaching adulthood. ARPKD also affects the liver, causing scarring and decreased liver function. Both parents must carry abnormal genes to pass on this form of the disease. If both parents carry a gene for this disorder, each child has a 25% chance of inheriting the disease.
Signs and symptoms of polycystic kidney disease can include:
- High blood pressure.
- Back or side pain.
- Blood in the urine.
- A feeling of fullness in the abdomen.
- Increased size of the abdomen due to enlarged kidneys.
- Headaches.
- Kidney stones.
- Kidney failure.
- Urinary tract or kidney infections.
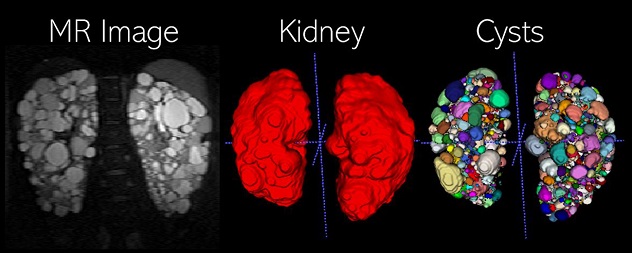 Abdominal MR Imaging in PKD
Abdominal MR Imaging in PKD
The kidney and cyst planimetry segmentations are obtained using two deep-learning models to accurately measure the total kidney volume and total cyst volume, respectively. Advanced imaging biomarkers are calculated from the individual (instance) cyst segmentation such as total cyst number and cyst-to-parenchyma surface area.
The PKD teams at Mayo Clinic's campuses in Florida, Arizona and Minnesota are each designated as a Center of Excellence for polycystic kidney disease by the PKD Foundation.
Additionally, Mayo Clinic Children's Center in Rochester, Minnesota, was awarded Pediatric Clinic designation by the PKD Foundation and offers diagnosis, care and treatment in a child-friendly environment for children with many types of cystic kidney disease. Each patient's PKD care team coordinates any necessary lab tests, imaging and referrals to other Mayo Clinic specialties.
"Our patients receive personalized treatment plans based on our state-of-the-art imaging and comprehensive genetic testing," says Dr. Hanna. "Mayo's multidisciplinary expertise in PKD allows us to treat each patient's unique complex needs depending on a variety of factors including their disease progression and pathogenic variants."
Research
There are two genes responsible for most cases of ADPKD, which have been named PKD1 and PKD2. PKD1 was discovered by geneticist Peter C. Harris, Ph.D., director of the Mayo Clinic Robert M. and Billie Kelley Pirnie Translational Polycystic Kidney Disease Center.
Dr. Harris has helped build a program at Mayo Clinic focused on identifying the many genetic variants that cause PKD and their effects on the course of the disease. In addition, Dr. Harris hosts the PKD Foundation-funded ADPKD Variant Database, which describes more than 2,000 variants of ADPKD genes.
It is still not exactly clear how pathogenic variants in PKD1 and PKD2 and recorded in the database cause PKD. However, studies using this data and published in the New England Journal of Medicine have enabled Mayo Clinic researchers to identify the first Food and Drug Administration-approved drug for treating the disease. That research traced the growth and expansion of kidney cysts back to a cascade of intracellular events known as the cyclic AMP signaling pathway. Cyclic AMP has been shown to play a role in all aspects of PKD, including increased growth of the cysts and abnormal fluid secretion into the cysts.
"Mayo Clinic is one of the major centers studying this disease in the United States," says Dr. Harris. "We have the resources — including research teams, clinical databases and biobanks — to continue investigating innovative therapies and treatments."
"Whether we can identify an appropriate clinical trial for a patient or whether they can benefit from an innovative therapy that originated from our research," continues Dr. Harris. "Our ultimate goal is to improve the lives of patients living with PKD — now and in the future."
For more information
Mayo Clinic Robert M. and Billie Kelley Pirnie Translational Polycystic Kidney Disease Center.
Polycystic kidney disease care at Mayo Clinic.
Torres VS, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. The New England Journal of Medicine. 2012;367:2407.
Torres VS, et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. The New England Journal of Medicine. 2017;377:1930.
Refer a patient to Mayo Clinic.