July 16, 2019
Two novel glial autoantibodies discovered in the past 15 years enable recognition of patient subsets with antigen-specific central nervous system inflammatory demyelinating autoimmunity manifesting as optic neuritis. Since introduction of live transfected cell-based assays, myelin oligodendrocyte glycoprotein immunoglobulin G (MOG-IgG) has emerged as a reproducible marker for a subset of patients with optic neuritis, aquaporin-4 immunoglobulin G (AQP4-IgG)-seronegative inflammatory central nervous system demyelinating disorders with neuromyelitis optica spectrum disorder (NMOSD)-like phenotype, and acute disseminated encephalomyelitis (predominantly in children).
Recent studies suggest the association of MOG-IgG seropositivity with recurrent optic neuritis attacks can lead to significant visual morbidity. Because there are few large studies of MOG-IgG-seropositive optic neuritis, however, the clinical phenotype is poorly defined.
To better define the clinical entity and anticipate visual outcomes, John J. Chen, M.D., Ph.D., a neuro-ophthalmologist at Mayo Clinic in Rochester, Minnesota, and a team of researchers conducted a multicenter, observational case series to determine the presenting signs and symptoms, radiologic abnormalities, accompanying neurological deficits, and visual outcomes of a large cohort of patients with MOG-IgG-seropositive optic neuritis. Study results were published in the American Journal of Ophthalmology in 2018.
Characteristics and visual clues
Researchers identified 87 patients seen at Mayo Clinic between 2001 and 2017, or elsewhere in 2016 and 2017, who had a clinically documented history of optic neuritis at any time and serum available that tested positive for MOG-IgG.
Patients were classified as having a single episode of optic neuritis, recurrent optic neuritis, chronic relapsing inflammatory optic neuropathy, neuromyelitis optica spectrum disorders-like phenotype, acute disseminated encephalomyelitis, multiple sclerosis, or "optic neuritis plus" for patients with additional neurological symptoms.
Patients' medical records were reviewed for presence of pain, fundus appearance at onset, visual acuity at the worst optic neuritis attack nadir and at last follow-up, number of attacks, other neurological symptoms, magnetic resonance imaging (MRI) findings, and immunotherapy and outcome.
Characteristics of the cohort include:
- Females comprised 57 percent.
- Median age at onset was 31 (range 2 to 79) years.
- Median number of optic neuritis attacks was 3 (range 1 to 8), median follow-up was 2.9 years (range 0.5 to 24 years), and annualized relapse rate was 0.8.
- Average visual acuity at nadir of worst attack was count fingers. Average final visual acuity was 20/30; for five patients (6 percent), average final visual acuity was less than or equal to 20/200 in either eye.
- Optic disk edema and pain each occurred in 86 percent of patients.
- MRI showed perineural enhancement in 50 percent and longitudinally extensive involvement in 80 percent.
- Twenty-six patients (30 percent) had recurrent optic neuritis without other neurological symptoms, 10 (12 percent) had a single episode of optic neuritis, 14 (16 percent) had chronic relapsing inflammatory optic neuropathy, and 36 (41 percent) had optic neuritis with other neurological symptoms (most often neuromyelitis optica spectrum disorder-like phenotype or acute disseminated encephalomyelitis).
- Only one patient was diagnosed with multiple sclerosis; that patient had a low MOG-IgG titer.
- Persistent MOG-IgG seropositivity occurred in 61 of 62 patients (98 percent).
- A total of 61 percent received long-term immunosuppressant therapy.
Bilateral grade 3 optic disk edema with peripapillary hemorrhages
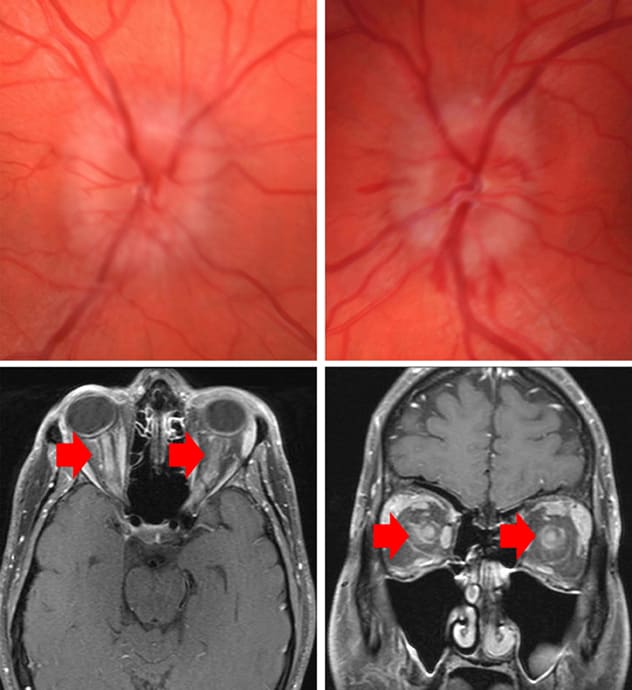
Bilateral grade 3 optic disk edema with peripapillary hemorrhages
Fundus images demonstrate bilateral grade 3 optic disk edema with peripapillary hemorrhages. Magnetic resonance imaging shows bilateral optic nerve enhancement with extension to the optic nerve sheath and surrounding orbital fat.
"Our team identified five major findings in our review of visual outcomes and characteristics in this cohort," says Dr. Chen. Those findings are as follows:
- The inflammatory course is diverse; in most cases optic neuritis is recurrent, with or without additional neurological features.
- Despite recurrence of attacks, most patients retain functional vision.
- Optic disk edema and bilateral disease are common.
- MRI evidence of optic nerve sheath and periorbital tissue involvement is common in the acute attack.
- MOG-IgG-positive neuroinflammation is a distinct entity from multiple sclerosis and AQP4-IgG-seropositive NMOSD.
"Our study indicates that MOG-IgG seropositivity predicts a relapsing inflammatory disease process with recurrent optic neuritis as a common feature. MOG-IgG positivity should be suspected if optic disk edema is moderate to severe at onset or if MRI shows optic nerve sheath involvement," says Dr. Chen. "Despite optic neuritis attacks being severe and recurrent, however, most patients retain good functional vision. It remains to be determined whether MOG-IgG serostatus in the remission phase of optic neuritis will predict future attacks."
For more information
Chen JJ, et al. Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis: Clinical characteristics, radiologic clues, and outcome. American Journal of Ophthalmology. 2018;195:8.