Dec. 15, 2018
Case 1
A 69-year-old man presented to the nephrology clinic for evaluation of rapidly progressing hypertension, hypokalemia and lower extremity edema over one month. He had a 20-year history of well-controlled hypertension on two medications. Despite multiple medication adjustments prior to evaluation, his hypertension remained poorly controlled. An endocrine work-up for secondary hypertension was performed, excluding pheochromocytoma and primary aldosteronism. However, the serum cortisol concentration after an overnight 1-mg dexamethasone suppression test was markedly abnormal at 88 mcg/dL, prompting referral to Endocrinology, Diabetes, Metabolism, and Nutrition at Mayo Clinic's campus in Rochester, Minnesota. In addition to hypertension, the patient complained of a two-month history of progressive weakness, muscle mass decline, severe fatigue necessitating wheelchair use, difficulty concentrating and mood changes. During this time, he had an episode of severe cellulitis requiring hospitalization. On physical examination, the patient did not have moon facies, supraclavicular pads, dorsocervical pad or striae.
Sagittal image of prostatic mass and multiple metastases
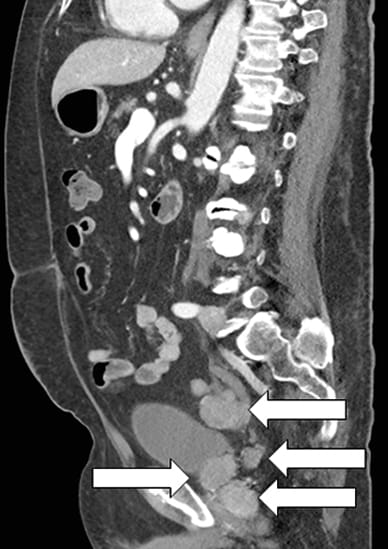
Sagittal image of prostatic mass and multiple metastases
Contrast-enhanced CT scan sagittal image showing prostatic mass and multiple metastases (arrows).
Laboratory study results for cases 1 and 2
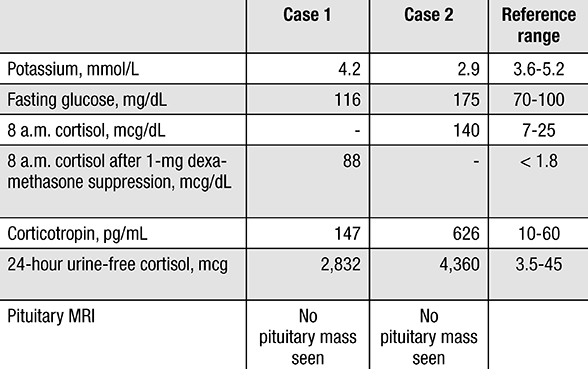
Laboratory study results for cases 1 and 2
Laboratory studies confirm severe ACTH-dependent hypercortisolism.
Prior to the Mayo Clinic evaluation, the patient had an abdominal CT scan performed for resistant hypertension. The scan revealed prostate enlargement with seminal vesicle invasion and multiple areas of intraperitoneal metastases. Prostate biopsy showed two distinct diagnoses of small cell carcinoma and prostate adenocarcinoma. Further laboratory studies confirmed severe corticotropin (ACTH)-dependent hypercortisolism, which was determined to be due to ectopic ACTH production from prostate small cell carcinoma.
Given the unresectable nature of the prostatic small cell carcinoma and the severe clinical and biochemical presentation, the decision for bilateral adrenalectomy was made and performed seven days after the initial abnormal dexamethasone suppression result. Perioperatively, the patient was initiated on glucocorticoid and mineralocorticoid replacement therapy.
Anticoagulation was started given the increased thrombosis risk, and antibacterial prophylaxis was initiated. One week after surgery, the patient was discharged to a skilled nursing facility to continue rehabilitation with a plan to follow up with Medical Oncology for management of the underlying malignancy.
Case 2
A 51-year-old woman presented to an outside facility for evaluation of acute abdominal pain. Imaging showed a 9.5-cm pancreatic mass, innumerable hepatic metastases and a bowel perforation. She had immediate abdominal surgery, and the intraoperative liver biopsy demonstrated a well-differentiated neuroendocrine tumor with positive immunohistochemical staining for gastrin. During her hospital stay, the patient was observed to have hyperglycemia, hypokalemia, muscle weakness and delirium. These new symptoms were concerning for abnormal cortisol secretion and further hormonal evaluation was performed. She was found to have severe hypercortisolism secondary to ACTH hypersecretion with a morning serum cortisol concentration of 140 mcg/dL (normal, 7 to 25 mcg/dL), serum ACTH concentration of 626 pg/mL (normal, 10 to 60 pg/mL) and 24-hour urine cortisol excretion of 4,360 mcg (normal, < 45 mcg). Her pituitary MRI showed no tumor.
Primary pancreatic neuroendocrine tumor and diffuse liver metastases
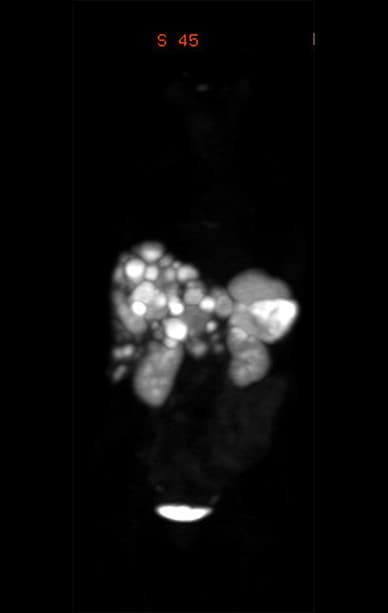
Primary pancreatic neuroendocrine tumor and diffuse liver metastases
Ga-68 DOTATATE PET scan showing the primary pancreatic neuroendocrine tumor and diffuse liver metastases.
The patient was transferred to Mayo Clinic for further management. Given the severity of her clinical and biochemical presentation as well as the unresectable nature of her tumor, the decision for bilateral adrenalectomy was made and was completed on the second day of her hospitalization. The patient's hospital course was complicated by Escherichia coli bacteremia (identified on admission blood cultures), postoperative hemorrhage requiring another surgical intervention and continued delirium. She was discharged 10 days after bilateral adrenalectomy with a plan to follow up with Medical Oncology for management of the metastatic neuroendocrine tumor. A postoperative gallium-68 (Ga-68) DOTATATE PET scan demonstrated the diffuse nature of her hepatic metastases.
Discussion
Ectopic ACTH production accounts for up to 20 percent of ACTH-dependent Cushing syndrome. Patients with ectopic ACTH secretion typically present with a much more rapid progression compared with those who have a pituitary-dependent Cushing syndrome. Frequently, patients with ectopic ACTH syndrome have muscle weakness, hypokalemia, hypertension and hyperglycemia that progresses over a short period of time. This rapid onset and marked production of ACTH, as well as the underlying malignancy, make significant weight gain and obesity less likely, which can cause a delay in the diagnosis of Cushing syndrome.
Although these patients may lack some of the more obvious clinical features of cortisol excess, they are at high risk of life-threatening complications such as infections and thrombosis. In the journal Cancer in 2011, clinical investigators at The University of Texas MD Anderson Cancer Center reviewed a 30-year experience with outcomes of 43 patients with ectopic ACTH secretion and found that 10 patients had infections and six patients had symptomatic venous thromboembolism, with two who died from pulmonary embolism. Follow-up of these patients showed a mortality rate of 62.8 percent with a median overall survival of 32.2 months.
In an article from the National Institutes of Health published in The Journal of Clinical Endocrinology and Metabolism in 2000, investigators showed that all patients who had ectopic ACTH production and severe infection had 24-hour urine-free cortisol excretion of more than 500 mcg (with most more than 1,000 mcg). Although medical therapy can decrease cortisol secretion or block its effect, these agents may take weeks for maximum effect, and even then may not be fully effective in correcting the clinical impact of hypercortisolism. Whereas, as documented in a report from Mayo Clinic published in Clinical Endocrinology in 2008, bilateral adrenalectomy is a relatively safe, prompt and extremely effective treatment option for ACTH-dependent Cushing syndrome that cannot be cured by resecting the source of ACTH.
These cases illustrate the importance of prompt recognition, diagnosis and treatment of ectopic Cushing syndrome. Both patients had 24-hour urinary cortisol excretion of more than 1,000 mcg — reflecting an endocrine emergency that required immediate treatment. In cases where a complete and prompt resection of the ACTH-secreting neuroendocrine tumor is not possible, bilateral adrenalectomy should be considered.
For more information
Ejaz S, et al. Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion: The University of Texas MD Anderson Cancer Center experience. Cancer. 2011;117:4381.
Sarlis NJ, et al. Cortisolemic indices predict severe infections in Cushing syndrome due to ectopic production of adrenocorticotropin. The Journal of Clinical Endocrinology & Metabolism. 2000;85:42.
Chow JT, et al. Bilateral laparoscopic adrenalectomy for corticotrophin-dependent Cushing's syndrome: A review of the Mayo Clinic experience. Clinical Endocrinology. 2008;68:513.